.jpg)
.jpg)
.jpg)
Чем характеризуются митохондриальные заболевания?
Какие диагностические критерии позволяют предположить развитие КМ, связанных с энергетической несостоятельностью митохондрий?
Какие препараты используются для коррекции кардиомиопатии при первичной митохондриальной патологии?
Кардиомиопатии (КМП) у детей относятся к тяжелым заболеваниям миокарда с непрерывно прогрессирующим течением и с высокой смертностью [1-3]. До последнего времени научные разработки по этой патологии были направлены на совершенствование методов диагностики заболевания, определение стадий клинического течения и поиск новых лекарственных препаратов для лечения недостаточности кровообращения [4-8].
Достижения медицинской науки за последние десятилетия в области медицинской генетики, биохимии и клинической морфологии помогли идентифицировать среди ранее недифференцированных патологических состояний новый класс митохондриальных заболеваний, обусловленных глубокими дефектами структуры и функции митохондрий [9, 10]. Развитие представлений о митохондриальной патологии позволило значительно расширить современные воззрения на патогенез заболеваний миокарда. Появилась возможность выделить особую группу метаболических митохондриальных КМП, что способствовало разработке дифференцированной специфической терапии.
Митохондриальные заболевания характеризуются полиорганностью поражения и проявляются в первую очередь поражением скелетных мышц, вовлечением в патологический процесс других органов и тканей (мозга, сердца, печени, почек) вариабельно, что приводит к гетерогенной клинической картине [9-14]. Описаны КМП при типичных митохондриальных заболеваниях: синдромах MERRF [11, 13, 14], MELAS [14-17], Кернса-Сейра [17, 18], митохондриальной миопатии [19]. Вместе с тем митохондриальная дисфункция может быть не только причиной развития полиорганной патологии, но и проявляться преимущественным поражением миокарда.
Экспериментальными и клиническими исследованиями последних лет установлено, что при так называемых «идиопатических» кардиомиопатиях происходят нарушения окислительного фосфорилирования, связанные со снижением активности митохондриальной электронно-транспортной системы вследствие мутации митохондриальной или ядерной ДНК. Существует предположение, что митохондриальная дисфункция может являться патогенетической основой кардиомиопатий, считающихся «идиопатическими» [14, 20-23]. Митохондриальные КМП могут быть определены как заболевания миокарда, сопровождающиеся структурными, количественными и функциональными нарушениями митохондрий либо комбинацией этих нарушений [14, 20-23]. Установлено, что основой формирования митохондриальных КМП являются мутации митохондриальной ДНК [20, 24-27]. Делеция, или точковая мутация, митохондриальной ДНК была выявлена в исследовании, проведенном Ито Т. (1992) [24] среди больных с гипертрофической или дилатационной кардиомиопатией и синдромом MELAS (митохондриальная миопатия, лактат-ацидоз, инсультоподобные эпизоды). Суомалайнен (1992) [26] приводит наблюдения за женщиной и ее сыном с дилатационной кардиомиопатией, обусловленной мутацией митохондриальной ДНК. Casali с соавт. (1995) [27] описывает новую мутацию митохондриальной ДНК в позиции 4300 tRNA(Ile), ассоциирующейся с ГКМП и материнским наследованием.
Мутации митохондриальной ДНК приводят к нарушению транспорта электронов вследствие патологии комплексов цепи дыхательных ферментов, что фенотипически проявляется КМП. Так, описаны КМП при дефиците цитохром С-оксидазы [28], снижении активности I и IV или II и III комплексов цепи дыхательных ферментов митохондрий [29], низкой активности пальмитоил-коэнзим А дегидрогеназы, связанной со снижением функции ацетил-коэнзим А дегидрогеназы длинных цепей [30].
В отечественной детской кардиологической практике митохондриальные КМП при жизни диагностируются недостаточно достоверно или остаются нераспознанными. Отсутствуют критерии дифференцированного подхода к обследованию больных с КМП для выявления митохондриальной недостаточности, не разработаны пути метаболической коррекции нарушений клеточной энергетики.
Мы наблюдали 16 больных с митохондриальными синдромами Кернса-Сейра (8 детей), Барта (1), MELAS (2), MERRF (1), гистиоцитарной КМП (1), карнитиновой КМП (2), с дефицитом II комплекса цепи дыхательных ферментов КМП (1 больной).
Программа исследования включала электрокардиографию, рентгенографию грудной клетки, суточное холтеровское мониторирование, допплер-эхокардиографию, позитронно-эмиссионную томографию (ПЭТ) сердца, неинвазивную оценку перфузии и метаболизма миокарда, морфологический анализ биоптатов скелетной мышцы, биохимическое определение в крови уровня лактата и пирувата. По показаниям проводились электромиография и электроэнцефалография.
Синдром Кернса-Сейра впервые описан Кернсом в 1946 году как заболевание с характерным клиническим симптомокомплексом: прогрессирующая наружная офтальмоплегия, пигментная ретинопатия, КМП с нарушением проводящей системы и развитием полного атриовентрикулярного блока. Более детальное изучение этого заболевания принадлежит Сейру, в связи с чем с 1956 года этот симптомокомплекс получил называние синдрома Кернса-Сейра. Пониманию природы синдрома способствовали молекулярно-генетические исследования и обнаружение мутаций митохондриальной ДНК [23, 31-34]. Впервые гистохимические и ультраструктурные изменения мышечных волокон в виде феномена «рваных» красных волокон (RRF), являющиеся важным критерием митохондриальной патологии, при синдроме Кернса-Сейра были обнаружены в 1972 году в работе Олсона и соавт. [35].
По нашим данным, при синдроме Кернса-Сейра при световой биопсии скелетных мышц феномен RRF определяется в 25% мышечных волокон. Обнаруживаются и другие маркеры митохондриальной патологии: субсарколемальные скопления липидов, гликогена, кальция. Электронная микроскопия выявляет нарушения структуры митохондрий: отсутствует часть митохондриальных крист, нарушено их обычное расположение (рис. 1).
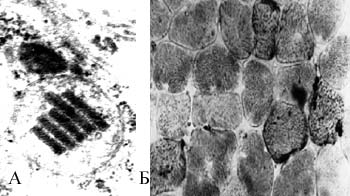 |
| Рисунок 1. Синдром Кернса-Сейра. А. Крупная митохондрия с деструкцией большинства крист и наличием сегмента параллельно лежащих, регенирирующих крист. ЭМ.х110 000 Б. Наличие субсарколеммальных полос продуктов реакции на СДГ в поперечно срезанных мышечных волокнах. Феномен RRF. Ув. 250 |
Клиническая манифестация синдрома Кернса-Сейра относится ко второму или даже третьему десятилетию жизни. Этот феномен объясняется тем, что суммарная функция митохондрий в течение длительного времени может оставаться удовлетворительной [36]. При этом, чем раньше проявляется заболевание, тем оно носит более генерализованный характер и имеет худший прогноз. Диффузные патологические изменения в митохондриях различных органов и тканей объясняют мозаичность клинической картины с вовлечением различных органов и систем. Отмечается задержка физического и полового развития. Изменения со стороны кожи проявляются ихтиозом с очагами гиперпигментации. Нарушения со стороны опорно-двигательного аппарата характеризуются вальгусной девиацией голеней и высоким сводом стопы. Наиболее выраженной является патология со стороны глаз. Характерны наружная офтальмоплегия с птозом различной степени выраженности, пигментный ретинит или пигментная дегенерация сетчатки. Птоз — наиболее типичный признак [10]. Один из наиболее частых симптомов — мозжечковая атаксия. У многих больных наблюдается умственная отсталость, однако степень снижения интеллекта варьирует от умеренно выраженной до прогрессирующей деменции. Описан случай инфаркта мозга у больного с синдромом Кернса-Сейра [37]. Нарушения со стороны эндокринной системы включают дефицит гормона роста, гипоганодизм, сахарный диабет, гипопаратиреоз, нарушение адреналового обмена [31, 38 ].
Изменения сердечно-сосудистой системы являются облигатной составляющей клинического симптомокомплекса синдрома Кернса-Сейра. Варианты и степень выраженности нарушений со стороны сердечно-сосудистой системы определяют тяжесть течения и прогноз синдрома Кернса-Сейра. Эти изменения в большинстве случаев затрагивают проводящую систему сердца. В исследованиях Мюллера с соавт. [39] обосновано преимущественное поражение проводящей системы сердца по сравнению с миокардиальными нарушениями у больных с синдромом Кернса-Сейра, большей представленностью мутаций митохондриальной ДНК в проводящей системе сердца в отличие от клеток сократительного миокарда. Так, частота мутаций митохондриальной ДНК у пациентов с синдромом Кернса-Сейра в клетках проводящей системы составляет 35-40% по сравнению с клетками сократительного миокарда — 10-20% [40]. Поражение проводящей системы сердца чаще всего характеризуется полным атриовентрикулярным блоком. В ряде случаев патология со стороны сердечно-сосудистой системы при синдроме Кернса-Сейра долгое время остается нераспознанной [41]. Иногда врачи проявляют серьезную озабоченность только при появлении полной атриовентрикулярной блокады, приводящей к резкой брадикардии, длительным паузам сердечного ритма с развитием синкопальных состояний — приступов Морганьи-Адамса-Стокса. Наличие полного атриовентрикулярного блока, не купированного своевременно имплантацией искусственного водителя ритма, является непосредственной причиной гибели этих пациентов [38, 41]. С другой стороны, даже имплантация искусственного водителя ритма не гарантирует благополучного прогноза. Так, Н. А. Белоконь с соавт. (1988) [42] обследовали ребенка с синдромом Кернса-Сейра, у которого на фоне полной атриовентрикулярной блокады частота сердечных сокращений уменьшалась до 28 в минуту, что сопровождалось развитием синкопальных состояний и потребовало имплантации искусственного водителя ритма. Однако, несмотря на это, больной умер через 9 лет от прогрессирующей сердечной недостаточности при удовлетворительном функционировании пейсмекера. Полная атриовентрикулярная блокада помимо угрозы возникновения приступов Морганьи-Адамса-Стокса приводит к возникновению миокардиальной дисфункции с последующим возможным развитием недостаточности кровообращения [38, 41]. Акэйк М. с соавт. (1997) подчеркивают, что высокий риск возникновения миокардиальной недостаточности на фоне полного атриовентрикулярного блока диктует необходимость имплантации пейсмекера типа DDD в ранние сроки, не дожидаясь опасных для жизни нарушений в проводящей системе сердца [43].
Под нашим наблюдением находилось 8 пациентов с синдромом Кернса-Сейра. Изменения со стороны проводящей системы не всегда манифестируют полным атриовентрикулярным блоком, возможно постепенное нарастание изменений со стороны проводящей системы сердца, что наблюдалось в 4 случаях. Степень поражения сердечно-сосудистой системы широко варьирует от дистального нарушения проведения по правой или левой ножке пучка Гиса до полной атриовентрикулярной блокады. Поражения проводящей системы сердца носят непрерывно прогрессирующий характер — они начинаются с дистальной блокады ножек пучка Гиса, распространяясь на атриовентрикулярный и синусовый узлы с развитием полной атриовентрикулярной блокады и синдрома слабости синусового узла.
Нарушения проводящей системы сердца при синдроме Кернса-Сейра могут сочетаться с изменениями в миокарде по типу КМП. Возможно формирование как дилатационной (3 из 8 наблюдений), так и гипертрофической симметричной необструктивной кардиомиопатии (2 из 8). Наиболее тяжелое поражение сердечно-сосудистой системы отмечено у девочки 15 лет с синдромом Кернса-Сейра. Наблюдалось диффузное прогрессирующее поражение проводящей системы сердца, начиная с дистальной двухпучковой блокады ножек пучка Гиса и транзиторной атриовентрикулярной блокады 1-й степени с последующим развитием блокады правой и левой ножек пучка Гиса до полной атриовентрикулярной блокады с длительностью периодов асистолии от 3 до 4 секунд и возникновением жизнеугрожающих синкопальных состояний — приступов Морганьи-Адамса-Стокса, что потребовало экстренной трансплантации искусственного водителя ритма. После имплантации ЭКС частота сердечного ритма составила 86 ударов в минуту. Нарушения проводящей системы сердца сопровождались развитием КМП, сочетающей как дилатацию полости левого желудочка, так и симметричную гипертрофию левого желудочка без обструкции. Особенности данного процесса следующие: развитие выраженной миокардиальной дисфункции, резкое снижение контрактильной способности миокарда и наличие обширных ишемических изменений в области переднебоковой стенки левого желудочка (деформация желудочкового комплекса по типу QS V1-5). Развитие миокардиальной дисфункции, снижение сократительной способности миокарда, распространенные ишемические изменения резко ухудшают прогноз течения синдрома Кернса-Сейра.
Синдром MELAS (митохондриальная миопатия — энцефалопатия — лактат-ацидоз, инсультоподобные эпизоды)
В основе патогенеза синдрома лежат точковые мутации митохондриальной ДНК. Мутация наиболее часто встречается в скелетной и сердечной мышцах, печени, почках, поджелудочной железе, мозжечке и коре больших полушарий [14]. Дебют заболевания вариабелен, наиболее часто это происходит между 6 и 10 годами. Экстракардиальными клиническими симптомами являются судороги, рецидивирующие головные боли, рвота, анорексия, непереносимость физических нагрузок, инсультоподобные эпизоды, деменция, миопатический симптомокомплекс, признаки периферической невропатии. При компьютерной томографии головного мозга выявляются зоны инфарктов, преимущественно в области гемисфер, что и обусловливает неврологическую симптоматику.
Под нашим наблюдением находились два пациента с синдромом MELAS. В одном из этих случаев наследственность отягощена. Старший ребенок умер в возрасте 10 лет (отмечались атрофия зрительного нерва, периодическая рвота, частые мигренеподобные головные боли, выраженная мышечная слабость, утомляемость, косоглазие), у матери — частые головные боли, непереносимость физических нагрузок. Дебют заболевания в том и другом случае относился к 8 годам. Первые симптомы — частые мигренеподобные головные боли, сопровождающиеся тошнотой и рвотой. Изменения неврологического статуса характеризовались мышечной слабостью, повышенной утомляемостью при физической нагрузке, нарушением походки: пошатывание при ходьбе, неуверенность при выполнении координационных проб. Впоследствии развились атаксия, нарушение речи (дизатрия, моторная дислалия), частые мигренеподобные головные боли, двустороннее снижение слуха, судорожные подергивания левой половины лица, сонливость. В 9 лет появились эпизоды тошноты, рвоты, снижения аппетита, усилились мигренеподобные головные боли. В 10 лет на фоне подъема температуры возник эпилептический статус, затем приступы стали чаще сопровождаться потерей сознания, клоническими подергиваниями левых конечностей и заведением левого глазного яблока кнаружи. Частота приступов в течение года нарастала, и в 12 лет возник инсультоподобный эпизод, в левых конечностях развился стойкий гемипарез. В одном случае отмечалось снижение остроты зрения. На ЭЭГ определялась пароксизмальная активность в виде групп острых волн с преимущественной локализацией в левой теменно-затылочной области. МРТ мозга: ишемические очаги в области головки хвостатого ядра и в белом веществе головного мозга кнаружи от внутренней капсулы слева. Нарушения зрения больше были характерны для девочки — отмечались снижение остроты зрения и частичная атрофия зрительного нерва. В 10 лет появились расходящееся косоглазие, птоз левого века, мелкоамплитудный горизонтальный нистагм, слабость конвергенции с обеих сторон. По данным аудиометрии, отмечалась двусторонняя нейросенсорная тугоухость.
 |
| Рисунок 2. ЭхоКГ — дилатация полости левого желудочка (КДДлж = 56 мм), снижение сократительной способности миокарда (фр. выброса 0,34) |
В одном случае изменения со стороны сердца характеризовались симметричной ГКМП (Тзслж = Тмжп = 11 мм при норме 7 мм). Отмечались нарушения ритма — эктопический предсердный ритм, суправентрикулярная экстрасистолия — по типу три- и квадригеминии, нарушение процессов реполяризации. Наиболее тяжелое течение заболевания отмечено у девочки 12 лет с дилатационной КМП. По данным ЭКГ, резко выраженная синусовая тахикардия, нарушение процессов реполяризации миокарда. Результаты ЭхоКГ выявили дилатацию левого желудочка (КДД = 56 мм при норме 40 мм), снижение контрактильной способности миокарда (ФВ = 0,34 при норме 0,60) (рис. 2). По данным позитронно-эмиссионной томографии, отмечена дилатация полостей левого и правого желудочков, резкое снижение метаболизма глюкозы в миокарде (накопление FDG18 стенками миокарда резко снижено во всем миокарде, невозможно выделить стенки и полости левого желудочка К FDG18= 0,6-1,1 при норме 2,5, реакция на нагрузку глюкозой отсутствует) на фоне нормальной перфузионной способности миокарда (рис. 3). Зафиксировано выраженное снижение активности цикла Кребса.
 |
| Рисунок 3. Нарушение метаболизма глюкозы в миокарде, по данным позитронно-эмиссионной томографии при синдроме Барта. Перфузия миокарда в пределах нормы. Снижение метаболизма глюкозы — накопление FDG18 снижено (К FDG18 =1,4 при норме 2,5) |
По данным биохимического анализа крови, в том и другом случае отмечалось повышение содержания молочной и пировиноградной кислот: содержание молочной кислоты в исходе — 2,8 ммоль/л, через час после нагрузки глюкозой — 3,0, через 3 часа — 3,2 ммоль/л ; содержание пировиноградной кислоты в исходе — 0,34 ммоль/л (норма 0,05-0,09), через час — 0,28, через 3 часа — 0,24 ммоль/л; рН — 7,38. По данным биопсии скелетной мышцы, размеры большинства мышечных волокон не изменены, имелись участки атрофии — до 20%, в них отмечены регионарные некрозы и локальные участки повышения регенераторной активности по интраволоконному типу. В 35% мышечных волокон при окраске на сукцинатдегидрогеназу определяется выраженный позитивный феномен RRF (рис. 3). При электронной микроскопии выявляются типичные участки пролиферирующих митохондрий. Гликоген и липиды распределены равномерно, в значительном количестве определяются конгломераты кальция. Цитохимическое исследование обнаружило снижение активности СДГ в лимфоцитах периферической крови — 13 ед. (норма 19-22 ед.).
Cиндром MERRF (миоклонус-эпилепсия и инфаркт мозга, RRF-волокна)
Сочетание миоклонус-эпилепсии с «рваными» красными волокнами скелетных мышц обнаружили в 1973 году Циарис с соавторами, но в качестве синдрома MERRF он был описан Фукухарой и его соавторами в 1980-м. В основе синдрома MERRF лежит точковая мутация в позиции 8344 в гене лизиновой tРНК. При этом снижается синтез белка, кодируемый митохондриальной ДНК, в первую очередь это касается субъединиц цитохромоксидазы. Заболевание наследуется по митохондриальному (материнскому) типу. Дебют MERRF вариабелен — от 3 до 63 лет. Экстракардиальными симптомами, доминирующими в клинической картине, являются миоклонус-эпилепсия, атаксия, деменция, потеря слуха и мышечная слабость. С этих же симптомов заболевание может манифестировать. При компьютерной томографии головного мозга выявляются множественные церебральные инфаркты. Именно эти изменения и обусловливают основную неврологическую симптоматику. В биоптатах скелетных мышц обнаруживаются типичные RRF. Ферментно-гистохимический анализ выявляет недостаточность цитохром С-оксидазы. При электронной микроскопии наблюдаются увеличение размеров митохондрий, их деформация, липидные включения [13].
Под нашим наблюдением находился ребенок с синдромом MERRF. Изменения со стороны сердца характеризовались симметричной гипертрофической КМП (Тзслж = Тмжп = 12 мм при норме 7 мм). Отмечался синдром Вольфа-Паркинсона-Уайта, что создавало предпосылки для возникновения жизнеугрожающих состояний.
Синдром Барта (кардиомиопатия с нейтропенией и гипостатурой)
В последнее десятилетие постоянно расширяется реестр митохондриальных болезней. В 1983 году П. Г. Барт и соавторы описали Х-сцепленный рецессивный фенотип, проявляющийся сочетанием скелетной миопатии, кардиомиопатии, задержки роста с нейтропенией в раннем возрасте. Наследование заболевания Х-сцепленное рецессивное. О митохондриальной природе заболевания говорят резко выраженные нарушения строения митохондрий мышечной, сердечной ткани и других органов. Заболевание возникает в раннем возрасте, на 5-7-м месяце жизни. Дети с данной патологией имеют низкий вес при рождении и в дальнейшем инфантильный соматотип (весо-ростовые показатели соответствуют 3-5 центилям), характерно отставание костного возраста от паспортного на 1-2 года. Заболевание манифестирует миопатическим синдромом. Изменения со стороны сердца могут характеризоваться как симметричной гипертрофической, так и дилатационной кардиомиопатией. Именно степень поражения сердечной мышцы определяет тяжесть и прогноз заболевания.
Нами наблюдался мальчик с синдромом Барта. Впервые изменения со стороны сердца были выявлены в 1,5 года в виде симметричной необструктивной гипертрофической КМП. По данным эхокардиографии, толщина межжелудочковой перегородки — 12 мм, задней стенки левого желудочка — 10 мм при норме 5 мм. При обследовании через год отмечено нарастание степени гипертрофии миокарда (толщина задней стенки левого желудочка — 13 мм, толщина межжелудочковой перегородки — 11,8 мм при норме 5,5 мм). На ЭКГ — повышение электрической активности левого желудочка, снижение процесса реполяризации в миокарде (рис 4).
 |
| Рисунок 4. Нарушение метаболизма миокарда, по данным позитронно-эмиссионной томографии при синдроме MELAS. Нормальная перфузия. Накопление FDG18 резко снижено. Снижение активности цикла Кребса. Снижение энергетического метаболизма вне связи с его кровоснабжением |
Основными клиническими симптомами являлись задержка физического (рост <3 центилей, масса <10 центилей) и психомоторного развития и повышенная утомляемость, отмечено умеренное отставание психоречевого развития. В неврологическом статусе определялись гипомимия лицевых мышц, периодическое поперхивание, нарушение произношения некоторых звуков. Выражены общая мышечная гипотония и гипотрофия, снижение силы мышц.
Сухожильные рефлексы снижены, больше в верхних конечностях. Походка изменена по типу утиной, ребенок с трудом поднимается по лестнице. В психологическом статусе отмечена легкая задержка психического развития — IQ 80 ед. Электронейромиография: повышение числа полиморфных потенциалов, диффузные изменения нервно-мышечного характера, миопатический тип кривой. Компьютерная томография головного мозга — картина «пустого» турецкого седла. Содержание в крови тиреотропного гормона и гормонов щитовидной железы было в пределах нормы. При проведении пробы с клофелином дефицита соматотропного гормона не обнаружено. Рентгенография трубчатых костей выявила остеопороз. Лейкоцитарная формула по типу нейтропении. Проведенное биохимическое обследование ребенка выявило гипогликемию (2,0-3,12 ммоль/л при N = 3,3–5,7). Отмечалось повышение уровня молочной кислоты в сыворотке крови до 2,63 ммоль/л (при N = 1,0–1,7 ммоль/л), после нагрузки глюкозой содержание молочной кислоты повысилось до 4,16 ммоль/л, а также отмечался высокий уровень пировиноградной кислоты в крови (0,17-0,23 ммоль/л при N = 0,05–0,09 ммоль/л). Выявлено нарушение экскреции органических кислот, возникающее у больных с синдромом Барта как следствие недостаточности митохондриальных энергетических процессов. Наблюдается низкое содержание карнитина.
Митохондриальный генез патологии подтвержден данными биопсии скелетной мышцы. Ребенку были назначены следующие препараты: L-карнитин — 500 мг/сут, цитомак — 4,0 в/м 4 курса в год, коэнзим Q10 — 60 мг/сут, ноотропил — 0,4 г/сут. При своевременной заместительной терапии препаратами, улучшающими энергетический обмен в сочетании с диетой с низким содержанием жиров, были отмечены положительная динамика со стороны сердечно-сосудистой системы, уменьшение миопатического синдрома, улучшение показателей физического развития, уменьшение степени гипертрофической кардиомиопатии, нормализация толщины межжелудочковой перегородки (до 7,5 мм) и задней стенки левого желудочка (до 7 мм), а также снижение экскреции жирных кислот.
Карнитиновая кардиомиопатия
Было установлено, что КМП могут сочетаться с резким снижением уровня карнитина в митохондриях [44, 45]. В ряде случаев причиной ДКМП у детей может быть генетический дефект транспорта карнитина [46]. Изолированная карнитиновая КМП клинически проявляется с 3-5 месяцев, имеет плохой прогноз, смерть наступает внезапно вследствие метаболического стресса. Асистолия связана с тяжелой гипогликемией, возможна патологическая брадикардия, sinus-arrest из-за блокады выхода [45]. Клинически отличить карнитиновую КМП от дилатационной аутоиммунного генеза сложно. Дифференциальной диагностике помогают следующие клинико-инструментальные показатели. Обычно при дилатационной кардиомиопатии наблюдаются изолированная дилатация полостей без явлений гипертрофии и выраженная гипокинезия стенок левого желудочка. При карнитиновой КМП наряду с гипертрофией миокарда отмечаются дилатация левого желудочка и явления фиброэластоза эндокарда. Характерными изменениями на ЭКГ являются «гигантские» зубцы Т в левых грудных отведениях, превышающие зубец R желудочкого комлекса [47].
 |
| Рисунок 5. Карнитиновая ДКМП. ЭхоКГ. Высокий вольтаж QRS, смещение ST, высокий положительный двугорбый Т больше зубца R, гипертрофия желудочков КДДлж= 65 мм, атриомегалия, фракция выброса — 0,20, гипокинез и дискинез, митральная регургитация 2-3-й ст. |
Нами наблюдались 2 больных с карнитиновой КМП. Приводим одно из наблюдений. Больной А. 9 лет с карнитиновой недостаточностью, вызвавшей развитие дилатационной кардиомиопатии. В возрасте 7 лет стал жаловаться на повышенную утомляемость, слабость, потливость, появились жалобы на частые боли в животе, выраженный миопатический синдром. Тогда же отмечались признаки недостаточности кровообращения: одышка, сердцебиения, гепатомегалия. При рентгенологическом обследовании выявлено увеличение размеров сердца, в основном за счет левых отделов, КТИ до 56%. По данным ЭКГ, выявлено повышение электрической активности миокарда левого желудочка. В стационаре по месту жительства был поставлен диагноз: ДКМП. Назначено лечение: преднизолон, мочегонные, ККБ и поляризующая смесь. Однако терапия оказалась неэффективной, нарастали явления недостаточности кровообращения. Увеличивалась дилатация левого желудочка, границы сердца расширены влево до переднеаксиллярной линии; на ЭКГ — синусовая тахиаритмия, гипертрофия левых отделов сердца, резко выраженные ишемические нарушения в виде «гигантских» зубцов Т. На ЭхоКГ — расширение полости левого желудочка (КДДлж = 66 мм при норме 41 мм), снижение сократительной способности миокарда (ФВ = 0,32), симметричная гипертрофия стенок левого желудочка (Тзслж = Тмжп = 12 мм при норме 8 мм), легочная гипертензия. УЗИ брюшной полости: гепатомегалия, расширение печеночных вен. На основании выраженного миопатического синдрома, снижения толерантности к физической нагрузке, гепатомегалии, рефрактерной к применению дигоксина и мочегонных препаратов, изменений со стороны сердца в виде сочетания дилатации левого желудочка, снижения контрактильной способности миокарда с явлениями гипертрофии стенок левого желудочка, усиления сигнала от эндокарда и специфических изменений на ЭКГ в виде «гигантских» зубцов Т возникло подозрение о развитии карнитиновой кардиомиопатии (рис. 5). В целях верификации диагноза был определен уровень карнитина в плазме. Он был снижен до 18 ммоль/мл при норме 25 ммоль/мл. В связи с этим в стандартную терапию дигоксином и мочегонными препаратами был подключен L-карнитин (элькар раствор) из расчета 75 мг/кг. На фоне данной терапии состояние значительно улучшилось, уменьшились явления недостаточности кровообращения до 1-й степени, сократились размеры полости левого желудочка (КДДлж уменьшилась с 66 мм до 52 мм), увеличилась контрактильная способность миокарда (фракция выброса выросла с 0,32 до 0,49).
Гистиоцитарная кардиомиопатия
Гистиоцитарная кардиомиопатия впервые была описана Д. Вотом в 1963 году как «арахноцитоз» сердечной мышцы. Было установлено, что гистиоцитарная кардиомиопатия — это типичное клиническое проявление дефицита цитохрома-В [21]. Клиническая симптоматика заболевания проявляется с 3-недельного возраста, как правило, до 1 года, характеризуется внезапным возникновением тахиаритмий (желудочковой или наджелудочковой локализации), частым развитием фибрилляции желудочков. Чаще болеют девочки в соотношении 5:1. Несмотря на интенсивную терапию, больные погибают через несколько недель после начала приступов желудочковой тахикардии. При аутопсии обнаруживаются сочетание гипертрофии миокарда с дилатацией полости левого желудочка, фиброэластоз сердца, при световой микроскопии отмечаются необычные очаги «обесцвеченного» миокарда главным образом в желудочках, в субэндо- и субэпикарде, меньше интрамурально, реже изменения могут наблюдаться и в предсердиях, иногда группы трансформированных клеток встречаются в основании створок митрального, трехстворчатого и аортального клапанов. Эти очаги содержат удлиненные мышечные волокна, похожие на миофибрилы, так называемые гистиоцитоподобные пенистые или онкоцитарные клетки. В цитоплазме гистиоцитоидных клеток содержится большое количество липидов и гликогена. При электронной микроскопии в гистиоцитоидных клетках наблюдается большое количество митохондрий причудливой формы с низким содержанием цитохрома-В.
Приводим клиническое наблюдение больной 1 года 2 месяцев с гистиоцитарной кардиомиопатией. Изменения со стороны сердца характеризовались дилатационной и гипертрофической симметричной необструктивной кардиомиопатией, фиброэластозом эндокарда, синдромом Вольфа-Паркинсона-Уайта, пароксизмальной суправентрикулярной тахикардией.
Первая беременность матери протекала на фоне угрозы прерывания в 3-5 недель, выраженного токсикоза, постоянной гипотонии. Во время беременности мать дважды (в 4,5 мес. и в 7 мес.) перенесла грипп в тяжелой форме. На сроке 20 недель находилась на стационарном лечении в связи с выраженной анемией (Нв = 60 г/л). Роды преждевременные, на 36-й неделе, кесарево сечение из-за разрыва шейки матки 3-й степени, низко расположенной плаценты. Во время операции сильное кровотечение. Девочка родилась с массой 2450 г, длиной 46 см, закричала сразу, оценка по шкале Апгар 7/8 баллов. Были признаки морфофункциональной незрелости. Через неделю после рождения у девочки впервые зарегистрирован приступ пароксизмальной тахикардии с ЧСС 240 уд/мин, который протекал с признаками недостаточности кровообращения, длился около суток, купирован в/в введением изоптина. При обследовании выявлены значительное увеличение полостей сердца, снижение сократительной функции миокарда, увеличение печени до 4 см. По данным ЭхоКГ, выраженное расширение полости левого желудочка (КДДлж = 30 мм при норме 24 мм), снижение фракции выброса до 0,35, гипертрофия миокарда левого желудочка: Тмжп = Тзслж = 9 мм при норме до 4 мм). Кроме того, отмечалось усиление сигнала от эндокарда левого желудочка (фиброэластоз). На ЭКГ — феномен WPW, тип А. В приступе регистрировалась суправентрикулярная реципрокная тахикардия. Приступ купирован внутривенным введением изоптина. При выходе из приступа регистрировалась длительная (около 3 секунд) асистолия. В стационаре продолжались ежедневные приступы пароксизмальной тахикардии, которые далее купировались внутривенным введением АТФ. Получала гормональную и метаболическую терапию, сердечные гликозиды, мочегонные препараты. Сохранялась постоянная тахикардия с ЧСС 160 уд/мин, на ЭКГ феномен WPW. Эхокардиографически определялись дилатация полости левого желудочка (КДДлж = 32 мм при норме 24 мм), снижение сократительной способности миокарда (ФВ = 0,35 при норме 0,70) и симметричная гипертрофия миокарда левого желудочка (Тзслж = Тмжп = 9 мм при норме 4 мм), усиление сигнала от эндокарда, признаки недостаточности кровообращения НК IIБ степени. Для лечения назначены дигоксин, с кардиотрофической целью актовегин, в качестве антиаритмических препаратов кордарон, финлепсин. За 1,5 месяца лечения состояние девочки значительно улучшилось: сократились размеры полостей сердца (до верхних значений возрастной нормы), но сохранялась гипертрофия миокарда левого желудочка, исчезли признаки недостаточности кровообращения, на ЭКГ феномен WPW приобрел транзиторный характер. В возрасте 1 года 1 месяца проведена вакцинация ППМ. Через сутки у ребенка внезапно возникла многократная рвота. Впервые с трехнедельного возраста развился тяжелый приступ тахикардии с частотой более 240 уд/мин, девочка госпитализирована в реанимационное отделение. На ЭКГ зарегистрирована политопная желудочковая тахикардия. Наблюдались выраженная одышка, пастозность, постоянная тахикардия с ЧСС 160 уд/мин, глухость сердечных тонов, увеличение печени до +4 см. Была вновь назначена, противоаритмическая терапия: дигоксин, кордарон, финлепсин, мочегонные препараты фуросемид, триампур. Впервые возникло синкопальное состояние во сне — девочка резко закричала, затем обмякла, констатирована остановка дыхания и сердечной деятельности. После проведения реанимационных мероприятий сердечная деятельность восстановлена, но сохранялась политопная тахикардия. Через час вновь зарегистрирована остановка сердца, девочка в сопорозном состоянии. В течение 2 часов еще 5 раз отмечались остановка сердца, фибрилляция желудочков. Консервативное лечение и реанимационные мероприятия дали временный эффект. Экстренно переведена в ИСХИ им. Бакулева для повторной дефибрилляции и решения вопроса о восстановлении ритма хирургическим путем. При проведении внутрисердечного электрофизиологического исследования во время дачи наркоза в течение 5 секунд отмечалась асистолия с восстановлением приступа политопной желудочковой тахикардии с ЧСС 240 уд/мин. При картировании правого предсердия — наиболее ранняя зона активной стимуляции расположена в межжелудочковой перегородке. При попытке частой стимуляции купировать пароксизм тахикардии не удалось. Отмечались отдельные синусовые комплексы с восстановлением приступа. Медикаментозное тестирование лидокаина и новокаинамида не дало эффекта, достигнуто лишь небольшое урежение сердечных сокращений до 170 уд/мин. По данным ЭФИ, имела место полиморфная левожелудочковая тахикардия с ретроградной вентрикуло-атриальной диссоциацией, по-видимому, эктопического или триггерного характера. Преходящий синдром WPW, тип А. Ввиду полиморфности тахикардии, обилия очагов эктопии хирургическое лечение было признано бесперспективным. В отделении состояние девочки оставалось крайне тяжелым. Попытки навязать ритм от наружного кардиостимулятора и проводимые реанимационные мероприятия не привели к восстановлению сердечной деятельности. В результате некупируемого приступа фибрилляции желудочков сердца наступила смерть от острой сердечной недостаточности. Паталогоанатомический диагноз: Гистиоцитарная кардиомиопатия младенческого возраста. Субэндо- и субэпикардиальная трансформация миоцитов левого желудочка в гистиоподобные клетки, множественные очаги гистиоподобных клеток в интрамуральных отделах обоих желудочков. Очаговая жировая дистрофия миокарда левого желудочка. Фиброэластоз эндокарда левых отделов сердца. Миогенная дилатация полостей сердца с расширением фиброзных колец всех клапанов. Непрерывно рецидивирующая полиморфная левожелудочковая тахикардия, неоднократно переходящая в фибрилляцию желудочков, преходящий синдром WPW, тип А.
Гистологическое исследование миокарда выявило крупные комплексы гистиоцитоподобных пенистых клеток с обилием липидных включений в цитоплазме среди пучков нормальных кардиомиоцитов. Наличие липидов указывало на резкое снижение окислительной способности миокарда, на его жировую дистрофию и гипофункцию. Такое состояние свидетельствует о резком снижении активности митохондриальных ферментов и предположительно указывает на наличие врожденной первичной митохондриальной патологии. Таким образом, данное наблюдение демонстрирует развитие кардиомиопатии, сочетающей черты дилатационной и гипертрофической с фатальным нарушением сердечного ритма у ребенка с гистиоцитарной кардиомиопатией. Найденные морфологические изменения в виде большого содержания липидов и гликогена в сердечной мышце, изменения структуры митохондрий в совокупности с литературными данными о дефиците при этой патологии III ферментного комплекса цепи дыхательных ферментов митохондрий позволяют предположить, что данные изменения обусловлены митохондриальной патологией.
Кардиомиопатии при дефиците П-комплекса цепи дыхательных ферментов
Сукцинат-конзимQ-оксиредуктаза является важным ферментным комплексом как цикла трикарбоновых кислот, так и респираторной цепи митохондрий. Комплекс состоит из 4 полипептидов: 2 больших полипептида представляют сукцинатдегидрогеназу и 2 малых — b тип цитохрома. П-комплекс кодируется ядерной ДНК; наследование аутосомно-рецессивное, Х-сцепленное рецессивное или доминантное с неполной пенентрантностью (Ангелини, 1993).
Клинический фенотип дефицита П-комплекса характеризуется прогрессирующей офтальмоплегией, энцефаломиопатией, атаксией, миоклоническими подергиваниями, миопатией и/или энцефалопатией, вторичным дефицитом карнитина, лактат-ацидозом. Изменения со стороны сердца проявляются кардиомиопатией. В 1993 году Ангелини описывал двух братьев с митохондриальной миопатией и гипертрофической кардиомиопатией. У 25-летнего пациента на ЭКГ отмечались желудочковая тахикардия, фибрилляция предсердий, клиника отека легких. Синусовый ритм был восстановлен только на фоне применения верапамила. Отмечалась кардиомегалия. По данным эхокардиографии, обнаружилась необструктивная гипертрофическая кардиомиопатия с преимущественной гипертрофией межжелудочковой перегородки. Отмечались изменения опорно-мышечного аппарата в виде сколиоза, проксимального лордоза, слабости мышц плечевого пояса. Миография подтвердила миогенный паттерн. Второй брат 19 лет предъявлял жалобы на загрудинные боли. В возрасте 8 месяцев у него был отмечен эпилептический приступ. Эхокардиография выявила асимметричную необструктивную КМП, толщина межжелудочковой перегородки составляла 30 мм, задней стенки левого желудочка — 19 мм. В неврологическом статусе отмечены атаксия и гипотония мышц плечевого пояса. Электромиография подтвердила миогенный паттерн. У матери этих пациентов также выявлен миогенный паттерн, по данным электромиографии. Анализ mt-ДНК не выявил значимых изменений. При биопсии мышцы выявлены ragged-red волокна, отложения липидов. Структура митохондрий нарушена: дезориентация в расположении крист. Гистоферментный анализ биоптатов скелетной мышцы установил снижение активности сукцинатдегидрогеназы на 35% по сравнению с нормой и сукцинат-цитохромС-редуктазы на 47% по сравнению с контролем. Уровень карнитина был нормальным в первом наблюдении и сниженным до 62% от нормы во втором.
Под нашим наблюдением находился больной с гипертрофической симметричной кардиомиопатией, необструктивной формой со вторичной дилатацией левого желудочка на фоне выраженной миокардиальной дисфункции, с распространенным кардиосклерозом, рефрактерной сердечной недостаточностью НК Пб Ш ст., недиференцированным объемным образованием в полости левого желудочка (тромб), диффузными дистрофическими изменениями в печени, задержкой физического и психического развития, которые обусловлены системной митохондриальной патологией.
Мальчик родился массой 3400 г, длиной 52 см, закричал сразу с оценкой по Апгар 8/8. Раннее развитие без особенностей. Манифестация заболевания относится к 9 годам, когда появились жалобы на повышенную утомляемость, нарушения походки. Отмечались задержка физического и психического развития, головные боли. На фоне перенесенного ОРВИ отмечены резкое снижение физической активности, отсутствие аппетита, миопатический синдром, резкое снижение массы тела, состояние продолжало ухудшаться, появились симптомы недостаточности кровообращения П Б степени. На ЭКГ — признаки гипертрофии левого желудочка, блокада передней левой ножки пучка Гиса, ишемические изменения в миокарде. На ЭхоКГ — симметричная гипертрофия миокарда, митральная регургитация 2-й степени, дилатация полости левого желудочка, снижение сократительной способности миокарда (Тзслж = Тмжп = 28, КДД ЛЖ = 47 мм, ФВ = 0,40). Косвенные признаки легочной гипертензии — расчетное давление в полости правого желудочка 60 мм рт. ст., объемное образование в полости левого желудочка (тромб) (рис. 6). Присоединение выраженной миокардиальной дисфункции у ребенка с ГКМП привело к резкому снижению сократительной способности миокарда и развитию дилатационной кардиомиопатии. Учитывая инфантильный соматотип ребенка, отставание в физическом и психическом развитии, выраженную гепатомегалию, рефрактерную к проводимой терапии, миопатический синдром, было выдвинуто предположение о миторхондриальном генезе заболевания. В терапию на фоне мочегонных препаратов были включены капотен, предуктал, эссенциале, аплегин. На этом фоне состояние несколько улучшилось, уменьшилась тахикардия, одышка, печень сократилась до 4 см. Гистоферментный анализ биоптатов скелетной мышцы выявил значительное снижение сукцинат-цитохромС-редуктазы — на 43% по сравнению с контролем.
 |
| Рисунок 6. Аномалия 2-го комплекса цепи дыхательных ферментов. ЭхоКГ. Концентрическая симметричная гипертрофия миокарда Тзслж = Тмжп = 28, митральная регургитация 2-й ст., дилатация полости левого желудочка, снижение фракции выброса (ФВ = 0,40) |
Разработаны диагностические критерии, позволяющие предположить развитие кардиомиопатий, связанных с энергетической несостоятельностью митохондрий.
Экстракардиальными критериями являются:
- инфантильный соматотип (масса и рост ребенка соответствуют 3-5 центилям);
- мышечная слабость, выявляемая в покое или после физической нагрузки;
- снижение толерантности к физической нагрузке;
- нарушения зрения (птоз);
- снижение слуха;
- инсультоподобные эпизоды неясного генеза;
- периодическая (циклическая) нейтропения;
- стойкое увеличение печени, рефрактерное к лечению сердечной недостаточности на фоне приема сердечных гликозидов и диуретиков;
- увеличение уровня лактата и пирувата;
- повышенная экскреция органических кислот;
- снижение содержания карнитина в крови;
- ацидоз.
Особенностями кардиомиопатий, протекающих на фоне первичной митохондриальной патологии, являются:
- нарушение проводящей системы сердца;
- злокачественные, рефрактерные к антиаритмическим препаратам желудочковые аритмии у детей раннего возраста;
- сочетание дилатации полостей сердца и гипертрофии миокарда, преимущественно задней стенки, с проявлениями фиброэластоза эндокарда;
- выявление гипертрофической кардиомиопатии в раннем возрасте;
- семейный характер заболевания;
- "гигантские" зубцы Т на ЭКГ в левых грудных отведениях.
По вопросам литературы обращайтесь в редакцию


