

Наследственные болезни обмена (НБО) — обширный класс моногенных заболеваний, обусловленных нарушениями различных белков, выполняющих разнообразные функции в метаболизме клетки. Выделяют более 600 нозологических форм НБО, которые, в свою очередь, подразделяют в зависимости от пораженного метаболического пути на 22 подкласса. Суммарная частота НБО в популяции достаточно высока и составляет 1 на 3–5 тыс. живых новорожденных [1, 3]. БСльшая часть НБО, протекающих с поражением нервной системы, манифестирует в раннем детском возрасте и носит неуклонно прогрессирующий характер, приводя к тяжелой инвалидности и дезадаптации больных. Клиническая диагностика этих заболеваний представляет значительные трудности. Они связаны с малой распространенностью отдельных нозологических форм, отсутствием специфических симптомов на ранних стадиях заболевания (плохая прибавка веса, срыгивания, беспокойство, нарушения сна, мышечная гипотония/гипертония, слабость зрительного контакта, задержка психомоторного развития), выраженным клиническим полиморфизмом, наличием атипичных форм [2, 3].
До постановки правильного диагноза дети, как правило, наблюдаются в стационарах по поводу различных заболеваний: перинатальное поражение нервной системы, последствия внутриутробной инфекции, эпилепсия. Основными критериями, позволяющими заподозрить наличие заболевания из группы НБО, являются: дебют в определенном возрасте, прогрессирующая задержка психомоторного развития; судороги, плохо поддающиеся коррекции с помощью базовой антиэпилептической терапии. Кроме того, при некоторых болезнях происходит вовлечение в патологический процесс других органов и систем (гепато- и спленомегалия, поражение кожи и ее придатков) [2, 3]. Приоритетную роль в их диагностике играют биохимические и молекулярно-генетические методы исследования.
На протяжении последних 200 лет, вплоть до 30-х гг. ХХ в., выявление наследственного заболевания было равносильно приговору для больного и его семьи, а эмпирические попытки лечения пациентов с этими тяжелейшими недугами являлись безуспешными. В начале 1930-х гг. впервые в мире невролог и генетик С. Н. Давиденков, основываясь на собственном клиническом опыте и достижениях экспериментальной генетики, указал на ошибочность мнения о неизлечимости НБО. Однако отсутствие сведений о патогенетических механизмах развития этих заболеваний в тот период ограничивало возможности разработки методов их лечения, и все подобные попытки, несмотря на правильные теоретические установки, оставались длительное время эмпирическими [1]. В настоящее время благодаря успехам генетики в целом и существенному прогрессу теоретической и практической медицины многие НБО лечатся с помощью ферментзаместительной или специфической терапии [1, 18]. В частности, речь идет о таких заболеваниях, как фенилкетонурия, лейциноз (болезнь «мочи с запахом кленового сиропа»), гликогенозы, лизосомные болезни накопления (болезнь Гоше, болезнь Фабри), гомоцистинурия, недостаточность биотинидазы (НБ) и др. [11]. Поэтому вопросы ранней диагностики, лечения и профилактики этих недугов имеют не только большое медицинское, но и социально-экономическое значение [5, 17].
Недостаточность биотинидазы
Недостаточность фермента ведет к нарушению освобождения биотина из пищевого белка и рециркуляции эндогенного биотина. Активность биотинидазы в головном мозге человека крайне низка, поэтому для нормального функционирования нейронов необходимо достаточное и постоянное поступление биотина через гематоэнцефалический барьер. Уменьшение концентрации биотина при НБ на первых стадиях заболевания приводит к снижению активности пируваткарбоксилазы, что вызывает накопление лактата в головном мозге. Этот локальный лактат-ацидоз обусловливает появление в первую очередь неврологических симптомов [12]. Кетоацидоз является признаком продолжительной недостаточности биотина в организме и может не выявляться на начальных этапах заболевания. Причиной нейросенсорной тугоухости является накопление органических кислот, биоцитина и более крупных биотинильных белков [19]. Снижение уровня протективных жирных кислот, возможно, является причиной появления алопеции и кожной сыпи [11].
Клинические проявления
НБ манифестирует в среднем в 3–5,5 мес жизни. В редких случаях заболевание дебютирует в подростковом возрасте или на первой неделе жизни. Степень тяжести клинических проявлений заболевания зависит от уровня активности биотинидазы. Так, высокая остаточная активность фермента (от 25 до 30% от нормы) характерна для юношеских форм НБ [21]. При тотальной НБ (активность фермента менее 5% от нормы) заболевание манифестирует в первые месяцы жизни.
В раннем возрасте наиболее частыми начальными симптомами являются судороги: миоклонические, генерализованные или парциальные [9]. Иногда основным клиническим симптомом служит мышечная гипотония. В ряде случаев НБ манифестирует задержкой психомоторного развития, нарушениями ритма дыхания (дыхание по типу Куссмауля, ларингеальный стридор, апноэ), себореей, атопическим дерматитом, гнездной и/или тотальной алопецией, персистирующими конъюнктивитами, нейросенсорной тугоухостью [4, 8, 10, 13, 20].
У большинства пациентов имеет место сочетание неврологических и кожных нарушений. При дебюте заболевания в подростковом возрасте начальными симптомами являются мышечная слабость, спастические парапарезы, зрительные нарушения (снижение зрения, появление скотом, атрофия зрительных нервов) [14, 15].
В результате проведенных нами исследований за 2 года удалось выявить трех пациентов с точно верифицированным диагнозом: недостаточность биотинидазы. Диагноз был подтвержден биохимическими и молекулярно-генетическими методами: тотальное снижение активности биотинидазы в сыворотке крови, метаболического ацидоза, повышение уровня специфических органических кислот и концентрации лактата в крови и ликворе. При проведении ДНК-диагностики было установлено, что два пациента являются гомозиготными по мутации G98d7i3, а один — компаунд-гетерозиготой по частым мутациям G98d7i3/R538C. Известно, что наличие этих мутаций в любом сочетании сопровождается ранней и тяжелой манифестацией НБ [7].
В качестве иллюстрации приводим выписку из истории болезни одного из наблюдавшихся нами больных.
Больной Л. К., 5 мес, находился в отделении психоневрологии и эпилепсии РДКБ. Поступил с жалобами на судороги по типу серийных инфантильных спазмов, задержку психомоторного развития, снижение слуха, повышенную потливость.
Из анамнеза известно, что ребенок родился от 2-й беременности, протекавшей на фоне угрозы прерывания на 7–8-й неделе, роды первые, на 39-й неделе беременности, самостоятельные, вес при рождении 2900 г, рост 51 см, оценка по шкале Апгар — 9–10 баллов. Выписан из роддома на 5-е сутки жизни в удовлетворительном состоянии. Перенесенные заболевания: отит, конъюнктивит. С рождения отмечался тремор ручек и подбородка. В 2 мес появились серийные, флексорные, симметричные инфантильные спазмы до 10 раз в сутки. Проводился подбор противосудорожной терапии без явного положительного эффекта. На фоне гормональной терапии синактеном депо судороги стали реже, но сохранялись до 5–7 раз в сутки.
При поступлении — состояние средней тяжести. Кожные покровы чистые, сухие. Видимые слизистые бледно-розовые. Волосы на голове тонкие и редкие. Подкожная жировая клетчатка развита избыточно. Развернута нижняя апертура грудной клетки. Носовое дыхание свободное. Границы сердца не увеличены. Сердечные тоны ритмичные, шумов нет. В легких дыхание пуэрильное, хрипов нет. Живот мягкий, безболезненный, доступен глубокой пальпации. Печень: + 1 см из-под края реберной дуги, селезенка не увеличена. Наружные половые органы развиты по мужскому типу.
Неврологический статус: общемозговых и менингеальных симптомов нет. Сознание ясное. Форма головы округлая с выступающими лобными буграми. Окружность головы – 42 см.
Глазные щели — OD=OS, взор фиксирует, кратковременно прослеживает, зрачки округлой формы, симметричные, фотореакция сохранена, лицо симметричное, снижение реакции на звуковые раздражители, нистагм установочный в крайних отведениях, глоточные и небные рефлексы живые, дисфония, саливация не усилена. Голову не держит, поворачивается на бок с трудом. Мышечная гипотония, с умеренным повышением мышечного тонуса в дистальных отделах конечностей. Сухожильные рефлексы симметричные, живые. Патологические рефлексы — с двух сторон. На болевые раздражители реагирует. Задержка редукции безусловных рефлексов: ладонно-ротовой (+), Переса (+), Галанта (+).
В сознании, на осмотр реагирует адекватно, дифференцирует окружающих, успокаивается на руках у мамы.
При клиническом анализе крови и мочи отклонений от нормы не выявлено.
Ультразвуковое исследование внутренних органов: без патологии.
Электроэнцефалографическое исследование: типичных данных о наличии локальной, диффузной и генерализованной эпилептиформной активности за время данного исследования получено не было.
Компьютерная аудиометрия: регистрируется снижение слуха по типу нейросенсорной тугоухости I–II степени.
Офтальмоскопия: частичная атрофия зрительных нервов.
 |
| Рисунок 1. МРТ головного мозга пациента Л. К., 5 мес |
МРТ головного мозга: на сериях сагиттальных, фронтальных и аксиальных МР-томограмм в режимах Т1 и Т2 на фоне резко расширенных субарахноидальных конвекситальных пространств лобных, теменных, затылочных и височных областей и базальных цистерн отмечается хроническая субдуральная гематома обоих лобных и верхних отделов височных областей. Признаков кровоизлияний в вещество головного мозга не выявлено. Заключение: признаки задержки миелинизации мозгового вещества. Хроническая субдуральная гематома указанной области (рис.1).
Обследование в лаборатории наследственных болезней обмена веществ ГУ Медико-генетического научного центра РАМН показало снижение активности биотинидазы — 0,56 (4,40–12 нмоль/мин/мл).
К проводимой антиэпилептической терапии добавлен биотин в дозе 10 мг/сут. Спустя 2 нед после назначения биотина состояние улучшилось: ребенок стал держать голову и переворачиваться со спины на живот, судороги купированы, отметилась тенденция к нормализации биохимических показателей.
При повторном поступлении через 3 мес на фоне специфической терапии отмечалась положительная динамика: отсутствие судорог, ребенок хорошо переворачивается со спины на живот и наоборот, дифференцирует окружающих, улучшились слух, зрение и рост волос на голове, активный лепет, уровень лактата и органических кислот в норме (табл.).
Клинические проявления у всех обследованных нами больных достаточно сходные: начало болезни в первом полугодии жизни, сочетание неврологических нарушений с поражением кожи и ее придатков (алопеция, дерматит), судороги, не поддающиеся коррекции базовыми антиэпилептическими препаратами.
После установления диагноза всем пациентам был назначен биотин в суточной дозе 10 мг/сут, продолжалось проведение противосудорожной терапии. Через 1,5–2 нед от начала лечения отмечался парадоксальный эффект в виде регресса выявляемых до лечения расстройств. Судороги не возобновлялись (за исключением пациента Л. С., у которого диагноз был верифицирован на поздних стадиях болезни). Больные дети стали постепенно приобретать психомоторные навыки и проявлять реакцию эмоционального оживления на осмотр: улыбка, способность зрительного сосредоточения на лицах, улучшился мышечный тонус, отмечен рост волос (рис. 2). Столь высокая эффективность специфического лечения может объясняться нормализацией метаболических процессов в организме, что совпадает с данными литературы [6].
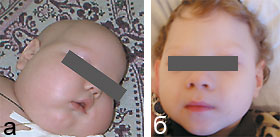 |
| Рисунок 2. Пациент Л. С. до терапии биотином (а) и после (б) |
Хотелось бы отметить, что от своевременной постановки диагноза и назначения специфической терапии зависит исход заболевания [16, 19]. Так, при ранней диагностике заболевания у больной П. Н. через 2 мес после начала патогенетической терапии психомоторное развитие соответствовало возрастной норме, тогда как у пациента Л. К. наблюдалась темповая задержка психомоторного развития. У одного из больных, несмотря на проведение специфического лечения, сформировался грубый неврологический дефицит (спастический тетрапарез, частичная атрофия зрительных нервов, нейросенсорная тугоухость, задержка психомоторного развития, редкие судороги), что может быть связано с наиболее ранним злокачественным течением заболевания (выраженный кетоацидоз и лактат-ацидоз) в исследуемой группе, а также его поздним выявлением (табл.).
Заключение
При выявлении у больного в первом полугодии жизни таких симптомов, как судороги, резистентные к проводимой антиэпилептической терапии, нарушения психомоторного развития, алопеция, дерматит, можно предположить наличие у пациента редкого недуга, относящегося к группе наследственных болезней обмена веществ, — недостаточности биотинидазы.
Применение специфической терапии биотином в дозе 10–30 мг/сут при данной патологии приводит к купированию судорог, регрессу неврологических расстройств, росту волос, восстановлению кислотно-основного равновесия и снижению уровня органических кислот в моче.
Исход заболевания зависит от своевременного установления правильного диагноза и назначения специфической терапии.
Литература
- Бочков Н. П. Клиническая генетика. - М., 2002. - С. 292.
- Темин П. А., Казанцева Л. З. Наследственные нарушения нервно-психического развития детей. - М., 2001. - С. 193-217.
- Aicardi. Jean Diseases of the Nervous System in Childhood 2nd Edition , 1998.
- Baumgartner E. R., Suormala T. M., Wick H. et al. Biotinidase deficiency: a cause of subacute necrotizing encephalomyelopathy (Leigh syndrome). Report of a case with lethal outcome// Pediatr Res. 1989; 26: 260-266.
- Lawler M. G., Frederick D. L., Rodriguez-Anza S. et al. Newbom screening for biotinidase deficiency: pilot study and follow-up of identified cases // Screening. 1992; 1: 17.
- Moslinger D., Muhl A., Suormala T. Molecular characterisation and neuropsychological outcome of 21 patients with profound biotinidase deficiency detected by newborn screening and family studies. // Eur J Pediatr. 2003; 162: 46-49.
- Pomponio R., Hymes J., Reynolds T. et al. Mutations in the human biotinidase gene that cause profound biotinidase deficiency in symptomatic children: molecular, biochemical, and clinical analysis //Pediatr. Res.1997; 42: 840-884.
- Rahman S., Standing S., Dalton R. N., Pike M. G. Late presentation of biotinidase deficiency with acute visual loss and gait disturbance. // Dev Med Child Neurol. 1997; 39: 830-831.
- Salbert B.A., Pellock J.M., Wolf B. Characterization of seizures associated with biotinidase deficiency. Neurology. 1993; 43: 1351-1355.
- Sander J.E., Malamud N., Cowan M. J. et al. Intermittent ataxia and immunodeficiency with multiple carboxylase deficiencies: a biotin-responsive disorder // Ann Neurol. 1980; 8: 544-547.
- Scriver C.R., Beaudet A. L., Sly W. S. // The Metabolic and Molecular Bases of Inherited Disease, New York: McGraw-Hill. 2001: 3935-3962.
- Suchy S. F., Rizzo W. B., Wolf B. et al. Fatty acids in biotin deficiency //Ann. NY Acad Sci. 1985; 447: 429.
- Suchy S. F., McVoy J.S., Wolf B. Neurologic symptoms of biotinidase deficiency: possible explanation //Neurology. 1985; 35: 1510-1511.
- Wastell H. J., Bartlett K., Dale G., Shein A. 1998. Biotinidase deficiency: a survey of 10 cases // Arch. Dis. Child. 1998; 63: 1244-1249.
- Wiznitzer M., Bangert B. A. Biotinidase deficiency: clinical and MRI findings consistent with myelopathy. // Pediatr Neurol. 2003; 29: 56-58.
- Wolf B., Heard G.S., Weissbecker K.A. et al. Biotinidase deficiency: initial clinical features and rapid diagnosis // Ann Neurol. 1985; 18: 614-617.
- Wolf B., Heard G.S. Screening for biotinidase deficiency in newborns: worldwide experience //Pediatrics. 1990; 85: 512-517.
- Wolf B., Heard G. S. Biotinidase deficiency //Adv Pediatr. 1991; 38: 21.
- Wolf B. Biotinidase Deficiency: New Directions and Practical Concerns.// Curr Treat Options Neurol. 2003; 5: 321-328.
- Wolf B., Grier R. E., Heard G. S. Hearing loss in biotinidase deficiency // Lancet. 1983; 10: 1365-1366.
- Wolf B., Grier R. E., Parker W. D. et al. Deficient biotinidase activity in late-onset multiple carboxylase deficiency // New Eng. J. Med.1983; 308:161.
С. В. Михайлова
Е. Ю. Захарова, кандидат медицинских наук
Е. С. Ильина, кандидат медицинских наук
А. С. Петрухин, доктор медицинских наук, профессор
РГМУ, PДКБ, МГНЦ, Москва
Приложения
-
- Диагностика и лечение недостаточности биотинидазы у детей раннего возраста - Таблица
Клинические проявления недостаточности биотинидазы и ответ на терапию биотином (по данным авторов)
Данные Пациент П. Н. Пациент Л. К. Пациент Л. С. Пол Ж М М Возраст дебюта заболевания 3 мес 2 мес 1,5 мес Возраст установления диагноза 9 мес 4 мес 4 мес Активность биотинидазы (норма 4,4-12 нмоль/мин/мл) 0,62 0,56 0,6 Проявления клинических симптомов* Симптомы До лечения На фоне терапии биотином До лечения На фоне терапии биотином До лечения На фоне терапии биотином Судороги + - ++ - +++ +/- Дерматит - - + - ++ - Атаксия + - - - - - Алопеция + - ++ +/- +++ - Конъюнктивит - - - - + - Ларингеальный стридор + +/- - - - - Мышечная гипотония/дистония + +/- ++ +/- +++ + Задержка психомоторного развития + - ++ +/- +++ + Атрофия зрительных нервов - - + +/- + + Нейросенсорная тугоухость - - ++ +/- ++ + Симптом "вишневой косточки" + - - - - - Сопор - - - - + - Метаболический кетоацидоз ++ - ++ - +++ - Лактат-ацидоз + - ++ - +++ - Уровень С5ОН** (норма < 1) 3,82 0,1 3,62 0,7 5,83 0,4 Катамнез через 6 мес после начала лечения Психомоторное развитие по возрасту Задержка темпов моторного развития Грубый неврологи- ческий дефицит * Степень проявления признаков: + — умеренная, ++ — выраженная, +++ — ярко выраженная.
** C5OH — 3-гидроксиизовалерил карнитин.
